

About ALSP
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is a fatal, rare, hereditary, and rapidly progressive neurodegenerative disease.1,2
ALSP by the Numbers
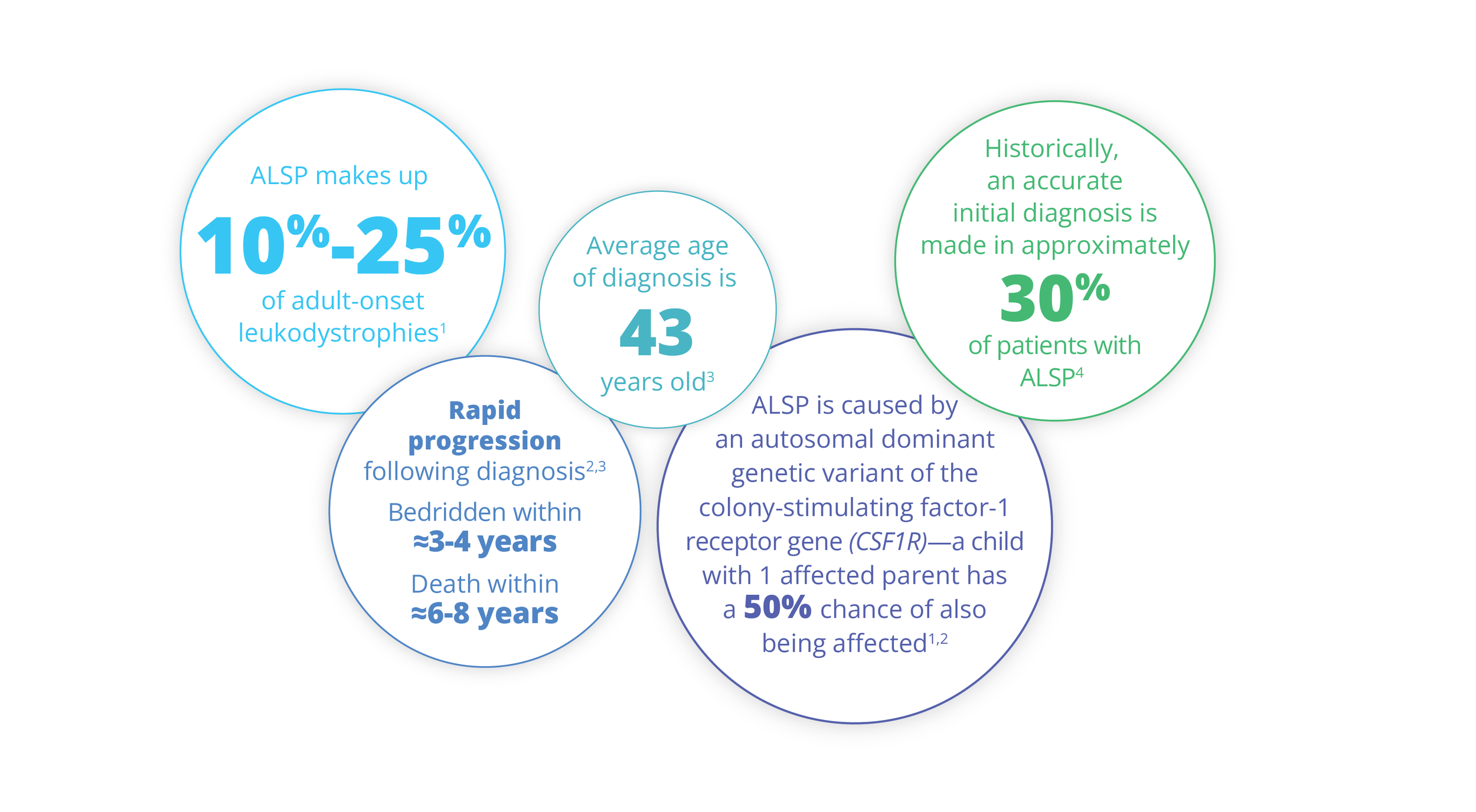


ALSP Overview
ALSP is a primary microgliopathy5
A microgliopathy is a hereditary loss of white matter in the brain caused by abnormal microglia, the resident sentinel immune cells of the brain, that cannot perform their critical role in development, maintenance, and repair of neural networks due to underlying gene mutations.5-7

Aβ, beta-amyloid; ApoE, apolipoprotein E; β-cat, beta-catenin; CSF1R, colony-stimulating factor-1 receptor; DAP, DNAX activation protein; ERK, extracellular regulated kinase; IL, interleukin; ITAM, immunoreceptor tyrosine-based activation motif; PI3K, phosphoinositide 3-kinase; Pyk2, proline-rich tyrosine kinase 2; SYK, spleen tyrosine kinase; TREM2, triggering receptor expressed on myeloid cells 2.
ALSP is a primary microgliopathy caused by loss-of-function mutations in the gene that encodes the colony stimulating factor-1 receptor (CSF1R).5,8 CSF1R, a transmembrane receptor that shares a common downstream signaling pathway with TREM2, is located on the cell surface of microglia and regulates cell survival, development, proliferation, and activation.2,8
Healthy microglial function
The ability of microglia to maintain the health of the central nervous system is dependent upon constant stimulation of the transmembrane receptor CSF1R to trigger signaling cascades resulting in microglial survival and proliferation, phagocytosis and elimination of damaged or foreign matter, and migration toward injured or inflamed tissue.5,6,9-11

Aβ, beta-amyloid; ALSP, adult-onset leukoencephalopathy with axonal spheroids and pigmented glia; ApoE, apolipoprotein E; CSF1R, colony-stimulating factor-1 receptor; DAP, DNAX activation protein; ITAM, immunoreceptor tyrosine-based activation motif; PI3K, phosphoinositide 3-kinase; SYK, spleen tyrosine kinase; TREM2, triggering receptor expressed on myeloid cells 2.
Microglial function in ALSP
In ALSP, loss-of-function mutations in the gene that encodes the CSF1R transmembrane receptor result in a dysfunctional or absent receptor, impairing the homeostatic functions of microglia that sense microenvironmental changes and respond by triggering survival, proliferation, phagocytosis, and motility.2,12 With fewer microglia available to develop, maintain, and repair brain tissue, the resulting white matter lesions show atrophy that leads to progressive cognitive and motor impairment and early death.2,9
ALSP is primarily inherited as an autosomal dominant genetic variant of the CSF1R gene.1,2

How the CSF1R mutation can be passed down from parent to children.
If one parent is affected with ALSP, each child has a 50% chance of inheriting the altered gene, resulting in ALSP.2
More than 100 CSF1R variants associated with ALSP have been identified, but no major genotype-phenotype correlations have been found to date.2
Although ALSP usually follows an autosomal dominant inheritance pattern, sporadic cases, including de novo mutations, have been reported.13

ALSP Clinical Course
Symptoms of ALSP typically appear in an individual’s 40s, but can begin in early adulthood or later in life (range, 18-78 years).3,14
ALSP is rapidly progressive14
By ≈3-4 years following diagnosis, patients may become wheelchair bound and caregiver dependent.3,14
By ≈6-8 years following diagnosis, patients may become bedridden and terminally ill.1-3,14,15 Death results most commonly from pneumonia or other infections.1,2,14
Estimated survival in patients with ALSP
In patients with ALSP, likelihood of survival decreases over time. An analysis of case reports from 291 patients with ALSP estimated a 75% survival rate after 3 years following diagnosis, 50% after 5 years, <25% by 10 years, and <5% by 30 years.16,17


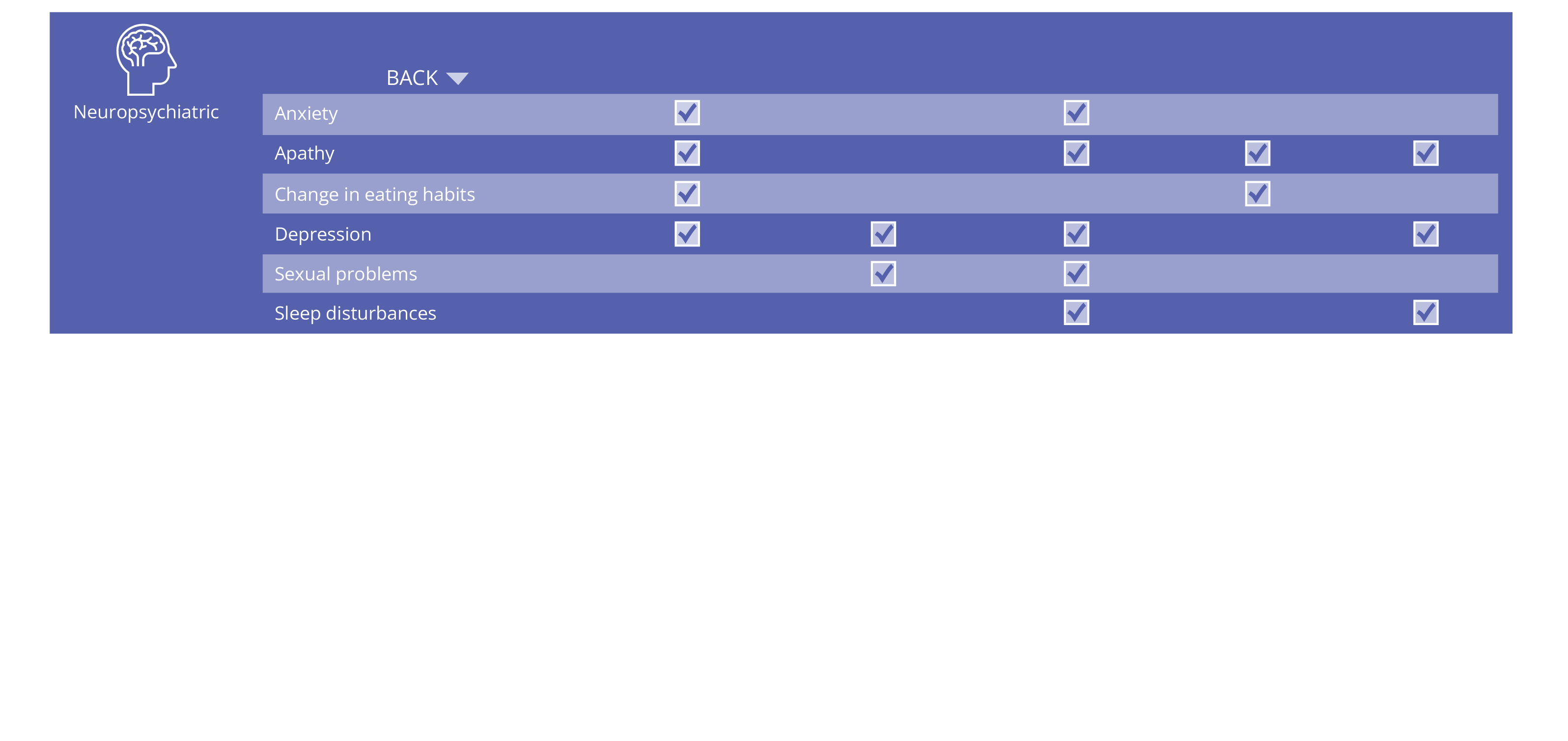
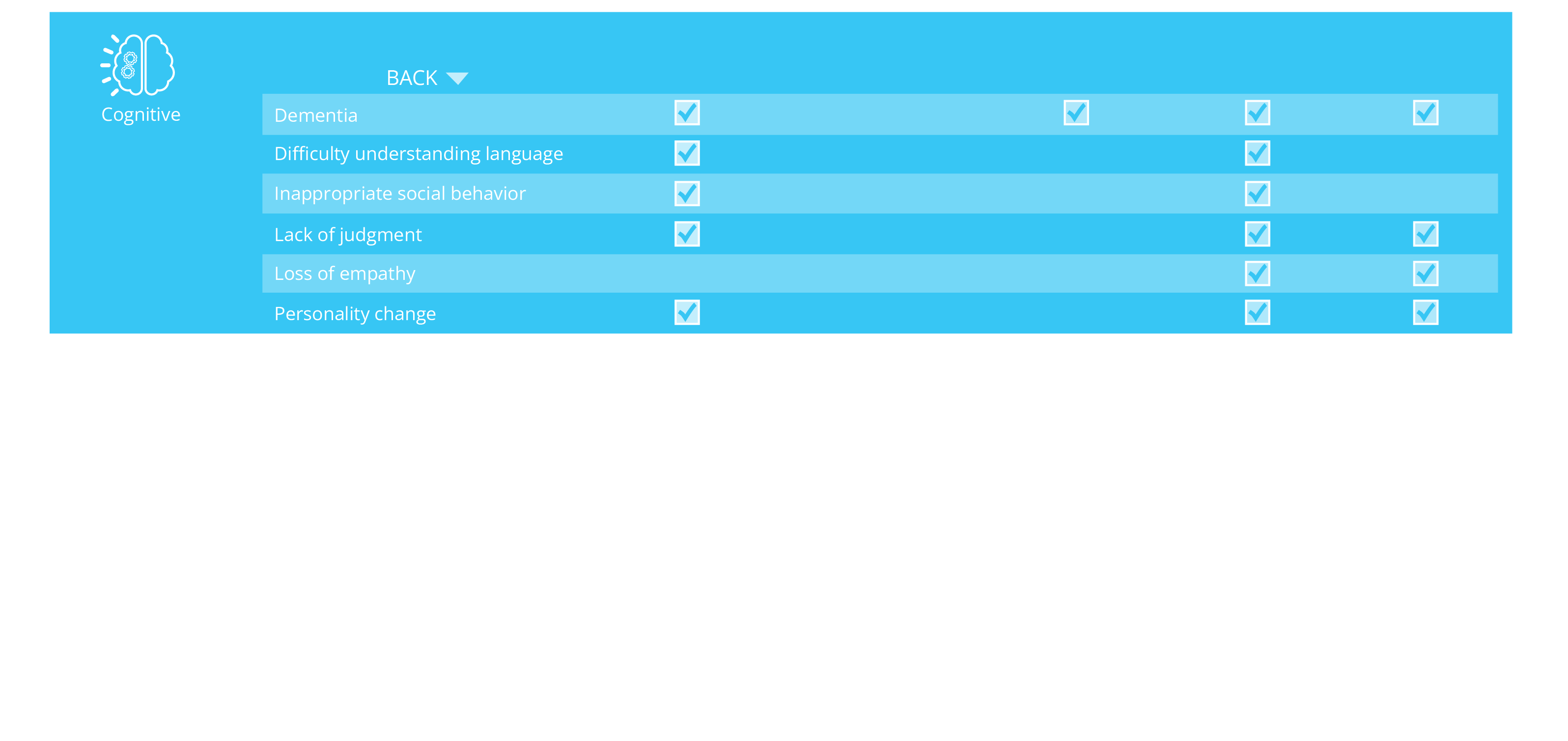
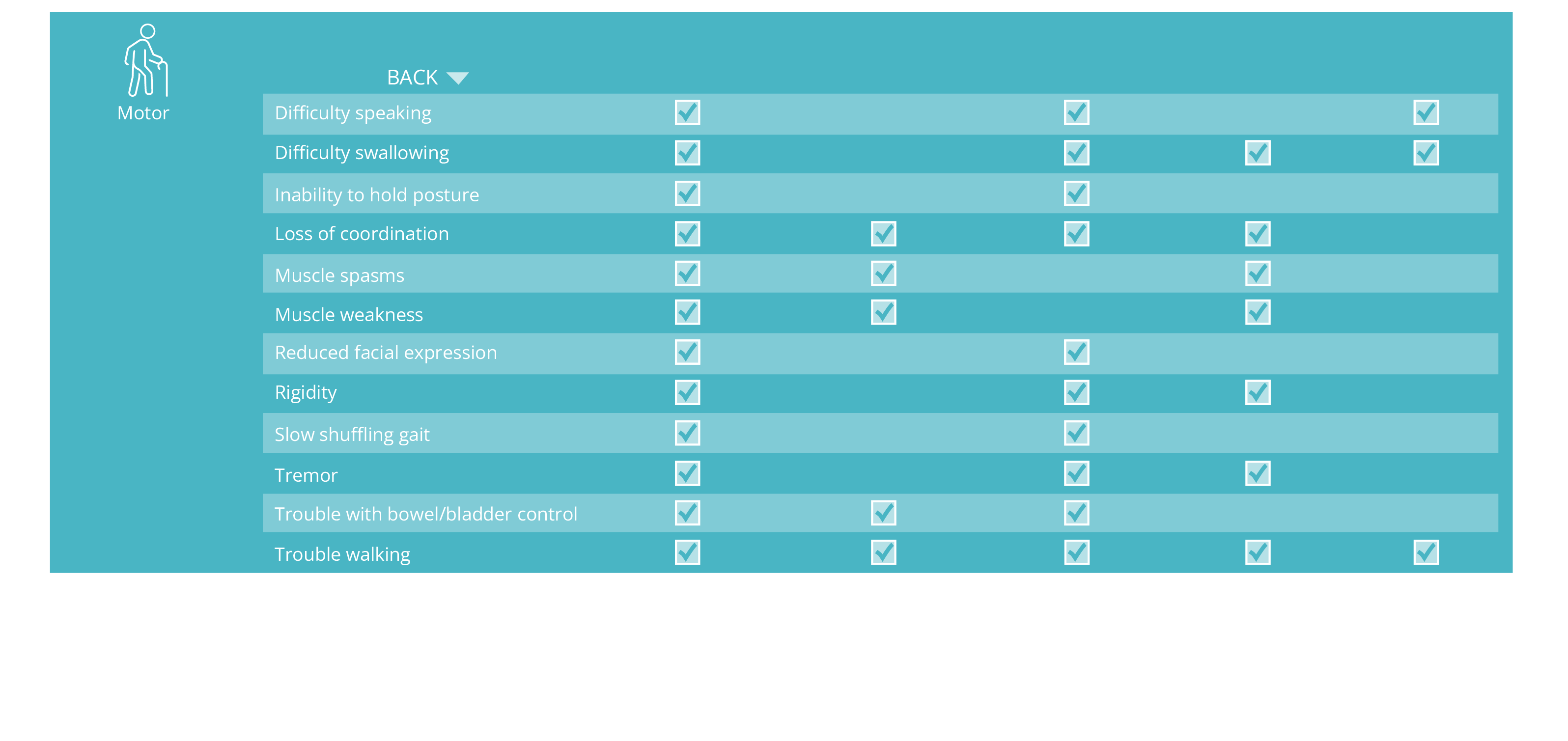
ALSP Signs and Symptoms
ALSP symptoms can be classified as cognitive, neuropsychiatric, sensory, or motor.1,2,13,14

Medical records from 57 patients with clinically or neuropathologically confirmed ALSP were retrospectively reviewed to characterize symptoms associated with ALSP presentation and disease progression18 :


Among patients with ALSP, cognitive impairment was the most common presenting symptom.18

Cognitive impairment was also the most prominent symptom of disease progression.18
Clinical characteristics
A literature analysis of 90 case reports published between 1980-2022 examined diagnosis and disease characteristics in 291 patients with ALSP confirmed by immunohistology, imaging, and/or genetic testing.4,17
Among patients who received a diagnosis at their initial presentation, only approximately one-third were accurately diagnosed with ALSP.4,17
Initial misdiagnosis with several other neurological and vascular disorders, including frontotemporal dementia, multiple sclerosis, and Alzheimer’s disease, was common.4,17 The differential diagnosis of ALSP is complex, and these data highlight a high potential for diagnostic delay and/or misdiagnosis due to the variety of presenting ALSP clinical and radiological characteristics that overlap with other neurological disorders.2,4,17,19
ALSP symptoms overlap with other neurological disorders







Histopathology
Swollen axonal spheroids and abnormally pigmented glia are characteristic of ALSP and may be visible by neuropathological examination of diagnostic brain biopsies, but presence is highly variable and subject to sampling bias and disease stage.25

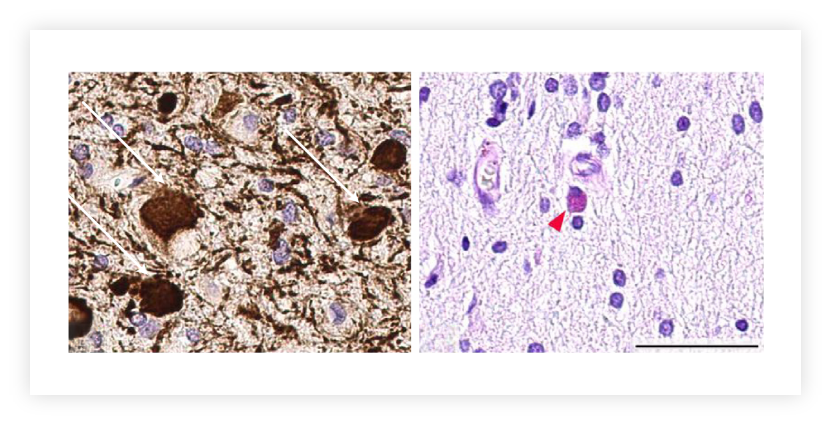
Images from reference 25, with permission under the Creative Commons Attribution 4.0 International (CC BY 4.0) license.
Immunostaining of biopsied subcortical white matter and cortical tissue shows frequent axonal spheroids (left; white arrows) and pigmented glial cells (right; red arrowhead).24
Scale bar, 50 μm.

Neuroimaging
Characteristic magnetic resonance imaging (MRI) features include confluent white matter changes, cerebral atrophy (corpus callosum), and hallmark stepping stone calcifications. 2,13,26-28 Preliminary data in patients with ALSP suggest that MRI measures of white matter lesion and brain volume are sensitive biomarkers of ALSP disease pathophysiology.29

Images from reference 30, with permission under the Creative Commons Attribution 4.0 International (CC BY 4.0) license.
MRI findings in a patient with ALSP show30:
(A) Confluent, slightly asymmetric frontoparietal predominant white matter atrophy
(B) Linearly arranged punctate and partly confluent hyperintense “deep white matter diffusion dots”
(C) Hyperintense involvement of the left corticospinal tract (white arrows), and
(D) Thinning of the corpus callosum.

If a patient presents with a combination of
Increased disease awareness and early genetic testing for CSF1R mutations can improve diagnostic accuracy.28,29,31 By increasing access to genetic testing, ALSPAware has the potential to reduce initial misdiagnosis of this disease while also providing appropriate disease management services for those living with ALSP.

ALSP Management
There are no FDA-approved therapies currently available for ALSP that target the underlying cause of the disease or slow its progression.1,2,13,14

In patients with ALSP, symptomatic therapies are currently used to manage motor, mood, and behavioral symptoms and to provide supportive care to maintain quality of life as the disease progresses.1,2,13,14
Allogeneic hematopoietic stem cell transplant (HSCT) has been used experimentally, but has not yet been tested in controlled clinical trials.

Psychological counseling is recommended for both patients and family members to cope with symptoms and progression of disease as well as potential risk of inheritance.1,2
References
1. NORD. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Accessed August 3, 2023. https://rarediseases.org/rare-diseases/adult-onset-leukoencephalopathy-with-axonal-spheroids-and-pigmented-glia/.
2. Papapetropoulos S, Pontius A, Finger E, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: review of clinical manifestations as foundations for therapeutic development. Front Neurol. 2022;12:788168.
3. Konno T, Yoshida K, Mizuno T, et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol. 2017;24(1):37-45.
4. Papapetropoulos S, Pontius A, Zappia S, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): initial misdiagnosis. Presented at EAN Annual Congress; June 25-28, 2022; Vienna, Austria. Poster EPV-631.
5. Berdowski WM, Sanderson LE, van Ham TJ. The multicellular interplay of microglia in health and disease: lessons from leukodystrophy. Dis Model Mech. 2021;14(8):dmm048925.
6. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441-468.
7. Fujita Y, Yamashita T. Neuroprotective function of microglia in the developing brain. Neuronal Signal. 2021;5(1):NS20200024.
8. Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200-205.
9. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458-3483.
10. Afridi R, Lee WH, Suk K. Microglia gone awry: linking immunometabolism to neurodegeneration. Front Cell Neurosci. 2020;14:246.
11. Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci. 2018;12:206.
12. Kempthorne L, Yoon H, Madore C, et al. Loss of homeostatic microglial phenotype in CSF1R-related leukoencephalopathy. Acta Neuropathol Commun. 2020;8(1):72.
13. Konno T, Kasanuki K, Ikeuchi T, et al. CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology. 2018;91(24):1092-1104.
14. Sundal C, Wszolek Z. CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. 2012 Aug 30 [Updated 2017 Oct 5]. In: Adam MP MG, Pagon RA, et al, eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2023.
15. Konno T, Yoshida K, Mizuta I, et al. Diagnostic criteria for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol. 2018;25(1):142-147.
16. Papapetropoulos S, Pontius A, Brennan M, et al. Survival analysis of patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): data from a systematic literature review of published case studies. Presented at AAN Annual Meeting; April 2-7, 2022; Seattle, WA. Poster P5-5.002.
17. Vigil Neuroscience, data on file.
18. Papapetropoulos S, Marsh A, Meier A, et al. Refining the phenotype of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), a commonly misdiagnosed autosomal dominant neurodegenerative disease. Presented at AAN Annual Meeting; April 22-26, 2023; Boston, MA. Oral presentation S49.010.
19. Makary MS, Awan U, Kisanuki YY, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: clinical and imaging characteristics. Neuroradiol J. 2019;32(2):139-142.
20. Johns Hopkins Medicine. Primary progressive multiple sclerosis. Accessed August 3, 2023. https://www.hopkinsmedicine.org/ health/conditions-and-diseases/multiple-sclerosis-ms/primary-progressive-multiple-sclerosis.
21. Mayo Clinic. Parkinson’s disease: symptoms & causes. Accessed August 3, 2023. https://www.mayoclinic.org/diseases- conditions/parkinsons-disease/symptoms-causes/syc-20376055.
22. Mayo Clinic. Frontotemporal dementia: symptoms & causes. Acessed August 3, 2023. https://www.mayoclinic.org/ diseases-conditions/frontotemporal-dementia/symptoms-causes/syc-20354737.
23. Cleveland Clinic. Frontotemporal dementia. Accessed August 3, 2023. https://my.clevelandclinic.org/health/diseases/ 21075-frontotemporal-dementia.
24. Alzheimer’s Association. 2023 Alzheimer’s disease: facts and figures. Accessed August 3, 2023. https://www.alz.org/ media/documents/alzheimers-facts-and-figures.pdf.
25. Lynch DS, Jaunmuktane Z, Sheerin UM, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. 2016;87(5):512-519.
26. Mickeviciute GC, Valiuskyte M, Platten M, et al. Neuroimaging phenotypes of CSF1R-related leukoencephalopathy: Systematic review, meta-analysis, and imaging recommendations. J Intern Med. 2022;291(3):269-282.
27. Rajagovindan R, O’Mara R, Meier A, et al. Radiological features of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and its longitudinal progression. Presented at AAN Annual Meeting; April 22-26, 2023; Boston, MA. Oral presentation P8-4.003.
28. Lynch DS, Wade C, Paiva ARB, et al. Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry. 2019;90(5):543-554.
29. Rajagovindan R, Matys B, Finger E, et al. A prospective natural history study of patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Presented at AAN Annual Meeting; April 22-26, 2023; Boston, MA. Oral presentation S49.008.
30. Lakshmanan R, Adams ME, Lynch DS, et al. Redefining the phenotype of ALSP and AARS2 mutation-related leukodystrophy. Neurol Genet. 2017;3(2):e135.
31. Meier A, Leahy L, Pontius A, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is commonly misdiagnosed as frontotemporal dementia (FTD), multiple sclerosis (MS,) Alzheimer’s disease (AD), or other adult-onset leukodystrophies. Presented at AAN Annual Meeting; April 22-26, 2023; Boston, MA. Poster P12-4.003.
32. Dulski J, Heckman MG, White LJ, et al. Hematopoietic stem cell transplantation in CSF1R-related leukoencephalopathy: retrospective study on predictors of outcomes. Pharmaceutics. 2022;14(12):2778.